Contributing to seq2science
Thanks for taking the time to contribute!!
The following is a set of guidelines and hints how to get started with contributing to seq2science.
I don’t want to read this whole thing i just have a question
Questions can be asked on the github issues page. Make sure to check if the question has been asked before in old issues and check the Frequently Asked Section (FAQ) in the docs if it’s there.
Reporting bugs
Bug reports can be made by making an issue on the github issues page.
Code/documentation
Outside of bugs/docs fixes, before you make a code contribution make sure to check in with us (the developers) if we are willing to support the contribution by making an issue first.
For an example on how to add to the documentation, see the example PR below.
Gitflow
We (try to) use the gitflow model to organise our parallel development. When making a PR, make sure to start from the develop branch and make a PR to develop branch.
Project design
Seq2science has grown to be a rather large project, with a complicated code structure. Before contributing to seq2science it is important to be up-to-date with the file and logic structure.
Documentation
The documentation can always be improved, and contributions are highly appreciated!
Apart from the auto-generated part of the docs, all the stuff relevant to the documentation is in the docs folder in the root.
The markdown files that make up the documentation are in docs/content.
tests
Seq2science runs tests on a Jenkins server after each push.
Everything related to the tests can be found in the tests folder in the root.
source code
The “source code” is split across multiple folders in the seq2science folder.
envs
In the seq2science/envs folder all the environments for each rule is stored.
When adding new rules make sure to add a new environment here.
imgs
These images are being shipped with seq2science. At the time of writing the only image that is in this folder is a Radboud University logo that is added to the MultiQC report.
rules
In seq2science/rules is where the magic happens.
Practically all rules and logic is in this folder. Rules are split across ‘logical’ topics, such as alignment, peak calling or downloading.
A workflow can thus easily include a couple of these files and link different topics.
All the files that start with configuration_ actually do not contain any rules.
These files are used to parse the samples.tsv and config.yaml.
These files are unfortunately pretty complex, but any time something big is changed, you probably have to change some stuff here.
schemas
This folder contains multple schemas.
It contains schemas to parse and extend the config.yaml at the start of the run, but it also contains e.g. the schema for the MultiQC report.
scripts
seq2science/scripts contains all the scripts that rules use to.
workflows
In seq2science/workflows are the different workflows.
Each workflow consists of a Snakefile, which defines what the total output is, which schemas are needed, and which rules.
In this folder is also an example samples.tsv and config.yaml, which is copied when using seq2science init.
util.py
Useful utility functions are deposited in this file.
important global variables
Global variables are variables that can be read from anywhere in the workflow.
Both Snakemake and Seq2science uses global variables to keep track of steps and samples.
Most global variables are declared in UPPER_CASE in seq2science/rules/configuration_generic.smk.
config is a dictionary with all the configuration stuff. Everything that you write in the config.yaml is in here. The config is parsed by the schemas (specified by the Snakefile) in
seq2science/schemas, and inseq2science/rules/configuration_.samples is the samples.tsv as a pandas dataframe, after parsing done in
seq2science/rules/configuration_. Unused columns are removed, and used columns with empty spaces are filled.ALL_SAMPLES is a list with all sample names from samples.
SAMPLEDICT is a dictionary containing information per sample that is not present in the samples.tsv file. In here is stored whether samples are SINGLE-end or PAIRED-end, and their corresponding SRR numbers. There are several sub-dictionaries of SAMPLEDICT:
ENA_SINGLE_END, ENA_PAIRED_END, SRA_SINGLE_END, SRA_PAIRED_END contain subsets of the samples,
RUN2DOWNLOAD contains the download link per sample run.
breps & treps, are dataframes with the technical and biological replicates merged (respectively). If you want to iterate over either of the technical replicates or biological replicates, or check what a sample belongs to, it is probably easiest to use these. There are several accompanying globals:
MERGED_TREPS is a list with technical replicates that contain >1 sample (will be merged during the workflow).
TREPS_FROM_BREP and BREP_FROM_TREPS are dictionaries that track which replicate contains which sub-replicates.
ALL_ASSEMBLIES is a list of assemblies used in the workflow.
PROVIDERS is a dictionary to keep track which assembly can be downloaded where.
HAS_ANNOTATION is a dictionary to keep track if an assembly has a gene annotation (or has one available for download)
CUSTOM_ASSEMBLIES is a dictionary to convert assembly names to custom assembly names, if you use custom assemblies.
ORI_ASSEMBLIES is a dictionary to convert custom assembly names to assembly names, if you use custom assemblies.
UCSC_ASSEMBLIES is a dictionary that denotes whether an assembly is already available on the UCSC trackhubs.
WORKFLOW is a string with the current workflow, as spelled in
seq2science/workflows(e.g. “chip_seq”).FINAL_BAM is a variable path to the (filtered and) coordinate sorted bam files with wildcards
{assembly}and{sample}.FINAL_BAI is the variable path to the matching bam index. We often use this to apply workflow-specific logic.
SEQUENCING_PROTOCOL contains the current workflow, spelled out properly (e.g. “ChIP-seq”).
Global wildcard constraints
Seq2science sets global wildcard constraints in seq2science/rules/configuration_generic.smk.
As wildcards are essentially greedy regex (.*), these constraints make sure we adhere to the naming convention of samples and assembly.
For example, one default wildcard constraint in seq2science is that the wildcard {replicate} can only match with
the technical_replicates column of the samples.tsv.
Example PR
If something in the documentation is wrong or unclear, please feel free to change it, no matter how small. Veteran FOSS (Free and Open Source Software) contributors already know how the process of suggesting changes to a project work (called a PR/Pull Request). However for people new to FOSS this relatively easy process can be a daunting task. Here we give an example of how to make changes to the documentation and propose them to us (the maintainers).
Make a github account
Make a “fork” of the repository
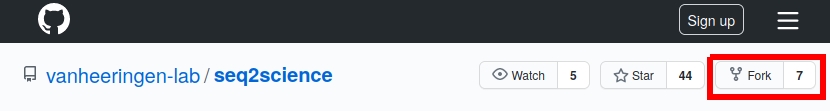
Now navigate on your “fork” to the documentation you want to change, for instance you could go to
docs/content/FAQ.mdto add a new question (and answer!).make some changes
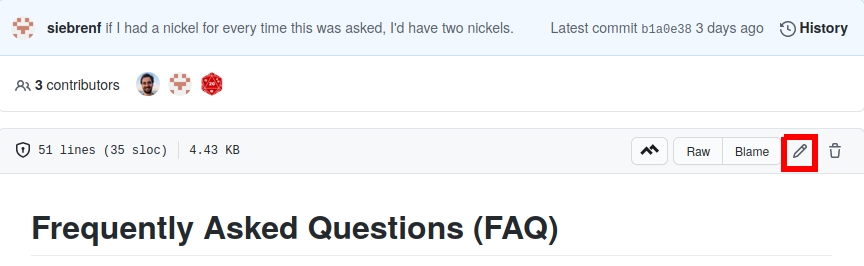
“commit” your changes to the
masterbranch`.Make a Pull Request (PR).
Wait for us to accept it…
See the changed docs online!





